The Study of Antibody Recognition©
This article is copyrighted by the Antibody Resource Page (https://www.antibodyresource.com/). It may be freely distributed so long as the ARP and its URL is included as its source. Educational institutions are especially encouraged to use this article as part of their immunology teaching curriculum.
The Function and Structure of Antibodies
The humoral immune system is a well designed fortress that defends its host against foreign invasion. The sentinels of this fortress are macrophages that continually roam the bloodstream of their host. When challenged by infection or immunization, macrophages respond by engulfing invaders marked with foreign molecules (antigens). This event, mediated by helper T cells, sets forth a complicated chain of responses that results in the stimulation of B-cells. These B-cells, in turn, produce proteins called antibodies which bind to the foreign invader. The binding event between antibody and antigen marks the foreign invader for destruction via phagocytosis or activation of the complement system.[1]
Five different classes of antibodies (or immunoglobulins) exist: IgA, IgD, IgE, IgG, IgM. They differ not only in their physiological roles but also in their structures. From a structural point of view, IgG antibodies are a particular class of immunoglobulins that have been extensively studied, perhaps because of the dominant role they play in a mature immune response. A considerable amount of what we know about the structure of antibodies comes from X-ray crystallographic [2] and immunoglobulin gene sequencing studies. [3] The structure of an IgG antibody is shown schematically in Figure 1. IgG antibodies are Y-shaped proteins composed of two heavy chains and two light chains that are joined by disulfide linkages. The IgG molecule can be broken down into two regions, the Fc and Fab. The Fc region, so called because it is the fragment of the IgG molecule that most readily crystallizes, is involved in effecting the physiological roles the antibody must play. Two identical Fab fragment are present at the ends of the "Y" in every IgG structure. The Fab region is named as such because it is the IgG fragment that contains the antibody binding site. The Fab region contains a region of highly conserved amino acids as well as a region of highly variable amino acids (Fv). These variable sequences are confined to 6 protein loops (or complementarity determining regions) that cluster together at the end of the Fab fragments to form a continuous hypervariable surface. It is this region that is responsible for the binding of foreign antigens.
Click for a labeled picture of an intact antibody.
Figure 1. Schematic drawing of an IgG antibody.
In order to perform their crucial role in the line of defense, antibodies must be extremely versatile. Indeed, on even a daily basis the immune system encounters a great variety of foreign substances (e.g. bacteria, viruses, toxins). As a result, antibodies must be extremely diverse to counter a large number of unexpected and unknown possibilities. Through a complex process of gene splicing, B-cells have been estimated to produce between 1 x 108 to 1 x 1010 IgG antibodies that differ in the composition of their binding sites.[4] This capacity to generate an enormous number of unique binding motifs makes antibodies a formidable line of defense.
Another needed attribute of antibodies is specificity. In order to distinguish between both self and a multitude of foreign species, antibodies need to have a highly discriminating method of recognition on the molecular level. This specificity is the result of the complementary nature of antibody binding. [5] This characteristic of antibody binding is the result of immunologically-tuned interactions (i.e. charge-charge, dipole-dipole, H-bonding, and Van der Waals) between the antigen and amino acid residues present in the antibody binding pocket. By taking advantage of the varied chemical properties of the 20 amino acids, the immune system is able to generate an array of antibody binding pockets that can accommodate the shape, charge, and hydrophobicity of seemingly any given antigen.
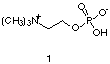 |
The complementary nature of antibody binding has been confirmed with the aid of X-ray crystallography. To date, a variety of crystal structures between Fab fragments and small molecules have been solved. In all cases, extensive use of intermolecular attractive forces are employed. For example, in the crystal structure between phosphocholine (1) and the antibody McPC603 [6] several well-placed amino acid residues can be seen to hold phosphocholine in the antibody binding pocket. In general, McPC603, makes critical use of coulombic interactions to recognize this antigen in direct response to the charged nature of the phosphocholine. For example, the negatively charged carboxylate group of an aspartic acid residue forms a salt bridge with the quarternized amine of the choline molecule. In addition, a guanidinium moiety from an arginine amino acid residue is observed to take advantage of both H-bonding and charge-charge interactions with the negatively-charged phosphate group present at the other end of the phosphocholine molecule. |
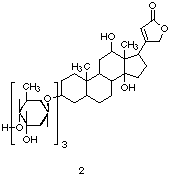
Also of interest is the crystal structure between Fab 26-10 and digoxin (2).
[7] This structure serves to demonstrate how hydrophobic effects can
play a critical role in antibody recognition processes. The digoxin
structure is composed of both a hydrophillic sugar unit and a
hydrophobic steroid unit. In this case, the hydrophilic sugar residue
is ignored; when digoxin is bound, the sugar dangles out of the
antibody binding pocket and into solution. Here, the high affinity
expressed by Fab 26-10 is the result of extensive contacts between the
steroid ring of digoxin and hydrophobic aliphatic/aromatic amino acid
side chains located inside a highly shape-selective binding pocket. |
|
Van der Waals contacts are used extensively by antibodies to conform to the shape of an antigen. For instance, in the crystal structure between the Fab 1F7 and a chorismate mutase transition state analogue (3), some 90% of the surface area (180 square angstroms) of the hapten is in tight contact with amino acid residues present in the antibody binding site. [8] While this example demonstrates how extensive this interfacial contact can be, it provides a somewhat incomplete picture of what is possible. Fab complexes with macromolecules, such as proteins, show that considerably more surface area is accessible to an antibody binding pocket to conform to the shape of an antigen. Interfacial contact between antibody and antigen have been shown to vary between 680 and 880 square angstroms for several complexes of antibody Fabs with lysozyme and with neuraminidase. [9] In these structures, all or almost all water has been excluded from the contact interface between the antibody binding site and the protein antigen. This exclusion of interfacial water, as evidenced in many complexes between Fabs and antigens, seems to be a common feature of antibody binding.
The high degree of complementarity exhibited by antibody binding also endows antibodies with high affinities for their antigens. For a mature immune response, antibody affinities typically fall in the range of 105 to 1012 M-1. [10] Recently, an upper ceiling for the affinity of a "normal" immune response has been proposed to be approximately 1010 M-1. [11] Two factors were used to derive this estimate. The first was the experimentally verified upper limit for on rates (105-106 M-1). [12] The second was an estimate of immunologically relevant off rates (10-3-10-4 s-1) related to T cell mediated processes.
Production and Isolation of Polyclonal and Monoclonal Antibodies
Two types of antibody samples can be used in the study of antibody-related phenomenon. The first type, polyclonal antibodies, [13] can be obtained by immunizing a mammal, such as a goat, sheep, mouse, or, most conveniently, a rabbit. After immunization, blood is removed (periodically, if desired) and the antibodies can be purified directly from the serum. The name polyclonal is derived from the Greek word for many (polys) and sprout (klon). As implied by the name, polyclonal antibodies originate (or "sprout") from a variety of B-cells that differ in the genetic material that encodes for antibody production. In a polyclonal sample, some of the antibodies will be specific for the antigen with which the animal was immunized. The remaining antibodies have been elicited from encounters with other foreign antigens that the animal has been exposed to throughout its lifetime.
The second type of antibody sample, the monoclonal antibody, [14] is derived from a more complex process. The technology associated with the production of monoclonal antibodies [15] is shown schematically in Figure 2. Here, a mammal, almost always an inbred mouse, is immunized with an antigen. After repeated immunizations, the spleen of the animal is removed. Because the spleen is responsible for B-cell production, the spleen cells contain the genetic information that gives rise to antibody production. Unfortunately, these spleen cells cannot be cultured. As a result, they are fused with "immortal" myeloma cells, so-called because of their ability to proliferate in vitro. The resulting fused cells, called hybridoma cells, are screened with a colorometric enzyme-linked immunoabsorbant assay (ELISA). [16] Use of this assay allows for the selection of hybridoma cells that produce antigen-specific antibodies. Because a given hybridoma cell is derived from a single B-cell, it produces a monoclonal antibody. Here, the prefix mono-, derived from the Greek word for single (monos), is used to indicate that a monoclonal antibody is derived from the genetic code of a unique B-cell. Once a single hybridoma line is selected, it is injected into a healthy mouse. Hybridoma cells, like myeloma cells, have the ability to produce tumors; consequently, after injection with a hybridoma line, a tumor grows inside the host mouse. When this tumor grows, it produces ascites, a fluid that is rich in monoclonal antibodies.
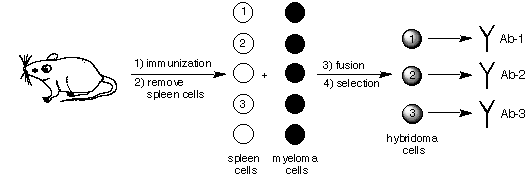
Figure 2. An outline of the procedure used to produce monoclonal antibodies. In this scheme three different antigen-specific antibodies produced via immunization are shown. Hybridoma cells that produce antibodies that are not specific for the antigen are easily screened out with an ELISA.
Once antibodies are produced, considerable care needs to be taken in their purification to avoid deleterious effects that may affect their study. The development of protein A and G-based chromatography has brought about considerable advances in the isolation of antibodies, in particular those of the IgG class. This chromatography utilizes a bacterial protein that binds specifically to the Fc region of IgG antibodies. [17] After a crude initial purification, serum or ascites can be passed through a chromatography medium that contains covalently attached protein A or G. At neutral to high pH, IgG antibodies are bound and unwanted biological substances, such as albumins and nucleic acids, can be washed through the column. When the pH is lowered, the bound IgG antibodies are eluted from the column. [18] Following subsequent neutralization and concentration, significant quantities of pure IgG antibodies can be obtained.
Polyclonal "Versus" Monoclonal Antibodies
Depending on the experimental situation, either a polyclonal or monoclonal antibody approach may be warranted. Each approach offers certain advantages. In the case of polyclonal antibodies, there are clear technical advantages. Polyclonal antibodies are inexpensive to produce relative to the cost of monoclonal antibody technology. In addition, large quantities of polyclonal antibodies (~10 mg/mL) can be produced from the serum of an immunized animal. Finally, high affinity polyclonal antibodies can be isolated merely 2-3 months after the initial immunization. This expeditious production facilitates their rapid study. There are also advantages to the use of polyclonal antibodies from a scientific perspective. Because polyclonal antibodies contain the entire antigen-specific antibody population, they offer a statistically relevant glimpse into the overall picture of an immune response. A similar viewpoint is considerably more difficult, if not impossible, using a monoclonal antibody approach.
On the other hand, monoclonal antibodies have certain advantages over polyclonal antibodies. Because of their immortal nature, hybridoma cells can be frozen, thawed, and recultured in vitro. As a result, for a given monoclonal line, there exists a constant and renewable source of antibodies for study. In addition, the defined composition of a monoclonal antibody allows for its chemical composition, on a molecular level, to be analyzed in detail. For example, X-ray crystallographic and gene sequencing methods can only be applied to monoclonal antibodies. This level of detail is particularly useful when studying mechanistic issues related to binding.
Chemical Approaches to Understanding Antibody Related Phenomenon
Many of the basic principles in the field of immunochemistry (the study of immunological processes on the molecular level) are now unreferenced material in the modern literature. Nevertheless, it is important to remember that these principles were established over half a century ago by chemists interested in unraveling the mysteries of antibodies. While commonplace today, the use of chemical methodologies to understand immunological phenomena early on in the field of immunochemistry was considered a revolutionary approach. One of the first attempts to study antibodies from the viewpoint of chemistry was taken up by Arrhenius. In 1907, Arrhenius published a series of his lectures entitled Immunochemistry: The Application of the Principles of Physical Chemistry to the Study of the Biological Antibodies. This text was of significance in that Arrhenius applied a mathematical approach founded in physical chemistry to explain various in vivo and in vitro phenomena related to antibody-antigen complex formation. In the preface of the book he wrote:
I have given to these lectures the title "Immuno-chemistry" and wish with this word to indicate that the chemical reactions of the substances that are produced by the injection of foreign substances into the blood of animals, i.e. by immunisation [sic], are under discussion in these pages. From this it follows also that the substances with which these products react, as proteins and ferments, are to be here considered with respect to their chemical composition. [19]
Arrhenius was the first to coin the term "immunochemistry". Here, for the first time in immunology, this branch of science mingled chemistry with biology to understand better the nature of antibodies. However, Arrhenius's text was most significant from a historical viewpoint. Perhaps because structure of known antigens (i.e. proteins and toxins) was extremely limited in the days of Arrhenius, the text is lacking in any detail that would leave one with a structural understanding of the nature between antibody and antigen.
Considerably more important was the work of Karl Landsteiner, the most recognized pioneer in the field of immunochemistry. In the 1930s, he investigated many of the fundamental principles of this field using an elegant molecular level approach to understanding antibody recognition. He made extensive use of immunoprecipitation, [20] which he called a "serological reaction", as a method to detect the binding of antibodies to antigens. In his authoritative text, The Specificity of Serological Reactions, he wrote:
For a considerable period of time after the discovery of serological phenomena and despite an abundance of observations, a method was yet wanting for the systematic investigation, along chemical lines, of specificity in serum reactions. It was indeed clear that serological reactions must somehow be dependent upon the chemical properties of the substances involved ... but insufficient chemical knowledge concerning the available antigens (proteins) ... made a closer analysis impossible. [21]
Landsteiner realized that while proteins were immunogenic (capable of inducing an immune response) the lack of structural knowledge of proteins made using them to study the chemical specificity of "serological reactions" impossible. He turned to the use of small molecules which had a defined chemical composition to study antibody specificity. Landsteiner was the first to systematically develop and chart the use of small molecules as probes of antibody recognition.
One important observation to come out of Landsteiner's investigations was that small molecules, in of themselves, are not capable of inducing an immune response. To circumvent this, he found that small molecules could be covalently attached to a carrier protein to engender an immune response. These small molecules, called haptens, contained a diazonium group, a functionality that readily forms a covalent bond with surface-bound lysines on a carrier protein. While this diazonium group strategy has been replaced with other methods of covalent linkage, [22] immunization with hapten-carrier protein conjugates is still practiced today to obtain hapten-specific antibodies.
Perhaps most significantly, Landsteiner found that antibodies were capable of discriminating subtle differences in molecular structure. He found that antibody recognition could be probed by quantifying the amount of immunoprecipition observed upon the addition of anti-hapten polyclonal serum to a solution of a small molecule-protein conjugate. If the small molecule bound to the antibodies present in the serum, precipitation occurred, as a result of extensive cross-linking of the antibodies to the small molecule-protein conjugate. Importantly, the protein attached to the small molecule of interest was different from that of the carrier protein coupled to the hapten. Consequently, any immunoprecipitation seen upon the addition of the anti-hapten antibodies was attributed exclusively to a binding event between the antibody and the small molecule. If the antibodies did not bind to the small molecule, precipitation was not observed. Landsteiner used any cross-reactivity that resulted in immunoprecipitation as direct evidence that there were structural similarities between the hapten and small molecule that the anti-hapten antibodies recognized. During his studies, he charted and quantified this behavior using a "+" to indicate the presence of considerable precipitate, a "?" to indicate an intermediate amount, and a "o" to indicate the absence of precipitate. Using this method, Landsteiner was able to "map" the composite binding site structures for a given set of hapten-specific polyclonal antibodies.
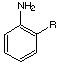
Landsteiner demonstrated that antibodies exhibited regioselective binding behavior. For example, as seen in the table shown in Figure 3, serum derived from immunization with 3-aminobenzenesulfonic acid exhibits very limited cross-reactivity with the corresponding ortho and para regioisomers. However, as would be expected, considerable precipitation is observed when the hapten itself is used. These results indicated that shape-selectivity plays a major role in antibody recognition.
| Antigens |
 ortho- |
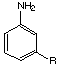 meta- |
para- |
| Aminobenzene sulfonic acid |
+ ? |
+ + ? |
? |
| Aminobenzene arsenic acid |
o |
+ |
o |
| Aminobenzoic acid |
o |
? |
o |
Figure 3. Reproduction of Table 21 taken from reference 21 showing the cross-reactivity between serum derived via immunization with a 3-aminobenzenesulfonic acid hapten and various small molecule (antigen)-protein conjugates.
With this same serum, Landsteiner also demonstrated that not only did the position of functional groups matter, but also their chemical composition. Very limited precipitation was observed when a carboxylic (-CO2H) or arsenic (-AsO3H2) acid was substituted for the sulfonic acid (-SO3H) group found in the hapten. When the other positional isomers of the carboxylic and arsenic acid substituted anilines were employed, absolutely no cross-reactivity was seen. These latter results indicated that molecular structures that deviated too far from the structure of the hapten were ignored by the anti-hapten antibodies.
Landsteiner used identical methodology to show that antibodies also exhibited both stereoselective and stereospecific binding. To demonstrate this , he used sera derived from the separate immunizations of the three isomers of tartaric acid. As seen in Figure 4, for a given tartaric acid isomer, there exists little cross-reactivity for one of the two other stereoisomers and no cross-reactivity for the remaining isomer. This experiment indicated that the shape and composition of a molecule were not the only factors recognized by an antibody; antibody recognition also seemed to take into account the spatial arrangement of functional groups around a given stereocenter.
| Antigens from: | |||
| l-Tartaric acid |
d-Tartaric acid |
m-Tartaric acid | |
| Immune Sera: |
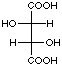 |
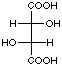 |
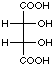 |
| l-Tartaric acid |
(+++) (+++) |
(?) (o) |
(+) (?) |
| d-Tartaric acid |
(o) (o) |
(++?) (+++) |
(+) (?) |
| m-Tartaric acid |
(?) (?) |
(o) (o) |
(+++) (+++) |
Figure 4. Reproduction of Table 23 taken from reference 21 showing the cross-reactivity patterns from sera derived via the separate immunization of the three stereoisomers of tartaric acid and the indicated molecule.
Landsteiner and his work has influenced several great scientific minds in field of immunochemistry. For instance, both Pauling and Pressman extended Landsteiner's work to develop the concept of an antibody binding pocket that was both complementary and shape-selective to its hapten. [23] This satisfyingly simple picture of the antibody binding site still serves as a model today. Indeed, modern X-ray crystallography has confirmed this conceptual model originally set forth by Landsteiner. The recent field of catalytic antibodies [24] is also rooted in the work of Landsteiner. Both Lerner and Benkovic, pioneers in this field, have payed homage to Landsteiner's legacy. [25]
Luminescent Probes of Antibody Structure
A variety of antibodies specific to, or elicited by, luminescent molecules are known. In some cases, such as antibodies to adriamycin, [26] water-soluble metalloporphyrins, [27] cascade blue, [28] and lucifer yellow, [29] little or no use of the luminescent properties of these small molecules to study antibody binding phenomena has been reported. However, there are three noteworthy examples of antibodies elicited to luminescent haptens with the sole purpose of better understanding the antibodies to which they bind:
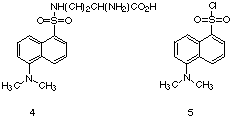 |
Parker utilized dansyl (DANS) derivatives, such as DANS-lysine (4), to investigate certain properties of polyclonal antibodies elicited via immunization with protein conjugates of 5. [30] While the DANS derivatives such as 4
exhibit some enhancement of emission in the presence of the control
protein, bovine serum albumin, the enhancement of emission observed
from anti-DANS antibodies was considerable greater. [31] Compared to
the emission seen for 4 in aqueous solution, the antibody
complexes of this fluorescent molecule are 150-200 times more emissive.
Using the enhancement of fluorescence as a measure of binding, Parker
was able to determine the average affinity of the anti-DANS polyclonal
antibodies. He demonstrated that the affinity of these antibodies for
anti-DANS-lysine increased as a function of the number of
immunizations. [32] |
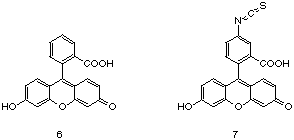
By far, the most extensively studied set of data for a luminescent antibody probe exists for fluorescein (6).
Indeed, the extensive number of studies that exists for this system has
made anti-fluorescein antibodies a paradigm for antibody binding in
general. [33] Both polyclonal and monoclonal antibodies have been
elicited via immunization with protein conjugates prepared with
fluorescein isothiocyanate (7). Antibody affinities have been reported to range from 104 to > 1012 M-1. [34] |
 |
One particular anti-fluorescein monoclonal antibody, 4-4-20, has been extensively studied and exhibits an association constant for fluorescein of 1.7 x 1010 M-1. [35] A crystal structure between the Fab of a monoclonal antibody, 4-4-20, and fluorescein has been solved. Similar to the binding motif between the Fab 26-10 and digoxin, the hydrophobic xanthonyl ring of fluorescein is buried in the antibody binding pocket and the hydrophilic, monoanionic carboxyl group extends out into solution. Extensive pi-pi interactions with tryptophan and tyrosine side chains present in the antibody binding pocket and fluorescein are also seen in this structure. [36]
In general, antibody binding of fluorescein results in quenching of up to 90% of the fluorescence relative to that of fluorescein in aqueous solution. Evidence that quenching by tryptophan residues present in or near the antibody binding pocket is responsible for this effect has been reported. [37] Use of this spectroscopic handle, pioneered by Voss, [38] has provided for a variety of elegant studies that include investigations of antibody-hapten dissociation and association rates, [39] heavy and light chain recombination rates, [40] and kinetic affinity maturation. [41] However, neither derivatives of dansyl chloride nor fluorescein have been used to investigate the molecular recognition of antibody binding, perhaps because of the limited availability of easily synthesized (or commercially prepared) derivatives. In addition, nearly all of the studies with these two types of luminescent antibody probes were conducted using steady-state fluorescence methodology. Time-resolved fluorescence techniques have not been applied to understand antibody binding phenomena related to these two systems.
Another class of molecules that has recently been exploited as luminescent antibody probes is ruthenium(II) metal complexes. More specifically, derivatives of Ru(II)(bpy)3, by far the most studied metal complex in the area of inorganic photochemistry [42], have been used. Several qualities of Ru(II)(bpy)3 facilitate its use as a probe of antibody binding phenomena. First, Ru(II)(bpy)3 is known to be both photochemically and structurally stable under aqueous conditions. [43] Furthermore, a variety of derivatives of Ru(II)(bpy)3 with a wide range shapes, sizes, charges, and electronic properties can be readily synthesized. [44] Finally, the MLCT transition for Ru(II)(bpy)3 absorbs at ca. 450 nm, distant from the UV-Visible region where antibodies strongly absorb. This factor allows for the selective excitation of this probe over any protein that is present.
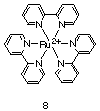 |
Ru(II)(bpy)3
has recently been used as a probe of polyclonal antibody
hetereogeneity. [45] Using standard time-resolved luminescence
methodology, the luminescence properties of a polyclonal antibody-bound
Ru(II)(bpy)3 metal complex indicated
that the sample in question was surprisingly homogenous from a
photophysical perspective. Despite the possibility of an extremely
diverse population of hapten-specific antibodies and the variety of
factors that can effect the excited state lifetime of the antibody
bound Ru(II)(bpy)3, this luminescent
probe exhibited simple photophysical behavior (i.e. a good fit to a
single-exponential lifetime). The underlying implication is that a
polyclonal response may not be as complex as previously thought.
Further work with other luminescent/fluorescent antibody probes should
help to shed light on the nature of the polyclonal antibody response. |
The molecular recognition properties of the monoclonal antibody AC1106, a murine antibody that exhibits cross-reactivity for derivatives of Ru(II)(bpy)3 and Ru(II)(phen)3, has also been studied. [46] Upon complexation, the studied metal complexes exhibited a significant enhancement of their excited state lifetimes. This enhancement of luminescence provided to be a convenient way to measure association constants between AC1106 and the metal complexes. Affinities for the metal complexes were shown to range from greater than 5 x 107 M-1 to less than 1 x 103 M-1. These values varied due to a variety of factors which included the shape and hydrophobicity of the metal complexes.
Conclusion
In order to play their physiological roles, antibodies are required to exhibit exquisite specificity and high affinity for foreign antigens. These properties have both fascinated and inspired chemists to better understand the unique properties of these immunological proteins. The molecular approach set forth by Landsteiner has strongly influenced how past and present researchers have studied antibody-related phenomena. A variety of techniques have been utilized to study antibody binding phenomena including immunoprecipitation, X-ray crystallography, and fluorescence spectroscopy. Future studies of the binding properties of antibodies will no doubt provide more clues about the complex operation of the immune system.
References
- Kuby, J. Immunology; W. H. Freeman and Company: New York, 1992.
- Davies, D. R.; Chacko, S. Acc. Chem. Res. 1993, 26, 421-427.
- Kabat, E. A.; Wu, T. T.; Reid-Miller, M.; Perry, H. M.; Gottesmann, K. S. Sequences of Proteins of Immunological Interest; National Institute of Health: Bethesda, Maryland, 1991.
- French, D. L.; Laskov, R.; Scharff, M. D. Science 1989, 244, 1152-1157.
- Pressman, D. In Molecular Structure and Biological Specificity, No. 2; Pauling, L.; Itano, H. A., eds.; American Institute of Biological Sciences: Washington, D.C., 1957; pp 1-17.
- (a) Padlan, E. A.; Davies, D. R.; Rudikoff, S.; Potter, M. Immunochem. 1976, 13, 945-949. (b) Padlan, E. A.; Cohen, G. H.; Davies, D. R. Ann. Inst. Pasteur/Immunol. 1985, 136C, 259-294.
- Jeffrey, P. D.; Strong, R. K.; Campbell, R. L.; Chang, C.; Sieker, L. C.; Petsko, G. A.; Haber, E.; Margolies, M. N.; Sheriff, S. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 10310-10314.
- Haynes, M. R.; Stura, E. A.; Hilvert, D.; Wilson, I. A. Science 1994, 263, 646-652.
- Davies, D. R.; Padlan, E. A. Ann. Rev. Biochem. 1990, 59, 439-473.
- Schultz, P. G.; Jacobs, J. W. In Environmental Influences and Recognition in Enzyme Chemistry; Liebman, J. F. and Greenburg, A., Eds.; VCH Publishers, Inc.: New York, 1988; pp. 305-312.
- Foote, J.; Eisen, H. N. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 1254-1256.
- (a) Northrup, S. H.; Erickson, H. P. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 3338-3342. (b) Raman, C.; Jemmerson, R.; Nall, B.; Allen, M. Biochemistry 1992, 31, 10370-10379.
- Garvey, J. S.; Cremer, N. E.; Sussdorf, D. H. Methods in Immunology; W. A. Benjamin, Inc.: Reading, Massachusetts, 1977.
- Goding, J. W. Monoclonal Antibodies: Principles and Practice; Academic Press: San Diego, 1986.
- (a) Kohler, G.; Milstein, C. Nature 1975, 256, 495-497. (b) K?hler, G.; Milstein, C. Eur. J. Immun. 1976, 6, 511-519. (c) Galfre, G.; Howe, S. C.; Milstein, C.; Butcher, G. W.; Howard, J. C. Nature 1977, 266, 550-552.
- Harlow, E.; Lane, D. Antibodies: A Laboratory Manual; Cold Spring Harbor Laboratory: Cold Spring Harbor, New York, 1988; pp 564-565.
- Bjorck, L.; Akerstrom, B. In Bacterial Immunoglobulin-Binding Proteins, Vol. 1; Academic Press: San Diego, 1990, pp 113-126.
- Jungbauer, A.; Tauer, C.; Reiter, M.; Purtscher, M.; Wenisch, E.; Steindl, F.; Buchacher, A.; Katinger, H. J. Chromat. 1989, 476, 257-268.
- Arrhenius, S. Immunochemistry: The Application of the Principles of Physical Chemistry to the Study of the Biological Antibodies; The Macmillan Company: New York, 1907, vii.
- Kabat, E. A. Structural Concepts in Immunology and Immunochemistry; Holt, Rinehart and Winston, Inc.: New York, 1968; pp. 53-62.
- Landsteiner, K. The Specificity of Serological Reactions; Harvard University Press: Cambridge, Massachusetts, 1945, p 156.
- (a) Erlanger, B. F. In Methods in Enzymology; Academic Press: San Diego, 1980; Vol. 70, pp 85-104. (b) Brinkley, M. Bioconj. Chem. 1992, 3, 2-13.
- (a) Pressman, D.; Brown, D. H.; Pauling, L. J. Am. Chem. Soc. 1942, 64, 3013-3020. (b) Pressman, D.; Swingle, S. M.; Grossberg, A. L.; Pauling, L. J. Am. Chem. Soc. 1944, 66, 1731-1738. (c) Pauling, L.; Pressman, D. J. Am. Chem. Soc. 1945, 67, 1003-1012. (d) Pressman, D.; Pardee, A. B.; Pauling, L. J. Am. Chem. Soc. 1945, 67, 1602-1606. (e) Pressman, D.; Siegel, M.; Hall, L. A. R. J. Am. Chem. Soc. 1954, 76, 6336-6341.
- Lerner, R. A.; Benkovic, S. J.; Schultz, P. G. Science 1991, 252, 659-667.
- Lerner, R. A.; Benkovic, S. J. Chemtracts 1990, 3, 1-36.
- Chien, Y.-H.; Levine, L. Immunochem. 1975, 12, 291-296.
- (a) Schwabacher, A. W.; Weinhouse, M. I.; Auditor, M.-T. M; Lerner, R. A. J. Am. Chem. Soc. 1989, 111, 2344-2346. (b) Keinan, E.; Sinha, S. C.; Sinha-Bagchi, A.; Benory, E.; Ghozi, M. C.; Eshhar, Z.; Green, B. S. Pure and Appl. Chem. 1990, 62, 2013-2019.
- Molnar, E.; Varga, S.; Jona, I.; Martonosi, A. Biochim. Biophys. Acta 1991, 1068, 27-40.
- Brandon, C.; Criswell, M. H. J. Histochem. Cytochem. 1991, 39, 1547-1553.
- Parker, C. W. In Experimental Immunology 1; Blackwell Scientific Publications; Weir, D. M., Ed.: Oxford, 1979, pp 18.1-18.25.
- Parker, C. W.; Yoo, T. J.; Johnson, M. C.; Godt, S. M. Biochemistry 1967, 6, 3408-3416.
- Parker, C. W.; Godt, S. M.; Johnson, M. C. Biochemistry 1967, 6, 3417-3427.
- Voss, E. W., Jr. Fluorescein Hapten: An Immunological Probe; CRC Press, Inc.: Boca Raton, Florida; 1984.
- Portmann, A. J.; Levison, S. A.; Dandliker, W. B. Immunochem. 1975, 12, 461.
- Mallender, W. D.; Ferreira, S. T.; Voss, E. W.; Coelho-Sampaio, T. Biochemistry 1994, 33, 10100-10108 and references therein.
- Herron, J. N.; He, X. M.; Mason, M. L.; Voss, E. W.; Edmundson, A. B. Proteins: Struct. Funct. Genet. 1989, 5, 271-280.
- Watt, R. M.; Voss , E. W., Jr. Immunochem. 1977, 14, 533-554.
- Lopatin, D. E.; Voss, E. W., Jr. Biochemistry 1971, 10, 208-216.
- Watt, R. M., Herron, J. N. & Voss, E. W., Jr. Mol. Immunology 1980, 17, 1237-1243.
- Watt, R. M.; Voss, E. W. J. Biol. Chem. 1979, 254, 7105-7110.
- Levison, S. A., Hicks, A. N., Portmann, A. J. & Dandliker, W. B. Biochemistry 1975, 14, 3778-3786.
- (a) Balzani, V.; Boletta, F.; Gandolfi, M. T.; Maestri, M. Top. Curr. Chem. 1978, 75, 1-64. (b) DeArmond, M. K.; Myrick, M. L. Acc. Chem. Res. 1989, 22, 364-370. (c) Meyer, T. J. Pure Appl. Chem. 1986, 58, 1193-1200. (d) Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A. Coor. Chem. Rev. 1988, 84, 85-277. (e) Kalyanasundaram, K. Photochemistry of Polypyridine and Porphyrin Complexes; Academic Press: San Diego, 1992; pp 87-164 and 339-361. (f) Roundhill, D. M. Photochemistry and Photophysics of Metal Complexes, Plenum Press: New York, 1994; pp 165-210.
- Brandt, W. W.; Smith, G. S. Anal. Chem. 1949, 21, 1313-1319.
- (a) Seddon, E. A.; Seddon, K. R. The Chemistry of Ruthenium; Elsevior Science Publishing Co. Inc.: New York, 1984; pp 414-475. (b) Shr?der, M.; Stephenson, T. A. In Comprehensive Coordination Chemistry, Vol. 4; Wilkinson, G., ed.; New York: 1987, pp 327-357.
- Shreder, K.; Harriman, A.; Iverson, B.L. J. Am. Chem. Soc. 1995, 117, 2673-2674.
- Shreder, K.; Harriman, A.; Iverson, B.L. J. Am. Chem. Soc. 1996, 118, 3192-3201.